PacBio Single Molecule Sequencing: Discontinued
GGBC is not currently offering PacBio Sequel II Sequencing
Click here for a list of recommended PacBio service providers.
General Information on Sequel II and Revio Sequencing Systems
The PacBio platform is based on Single Molecule, Real-Time (SMRT) technology. It is ideal for rapidly and cost-effectively generating high-quality whole genome de novo assemblies, full-length transcriptomes, cell type-specific isoforms (MAS-Seq) and long-read targeted amplicon sequences.
The new PacBio Revio system has all the capabilities of the Sequel II but with a higher throughput (up to 3X per SMRT-cell), better accuracy, and shorter run times for a lower cost per base than on the Sequel II.
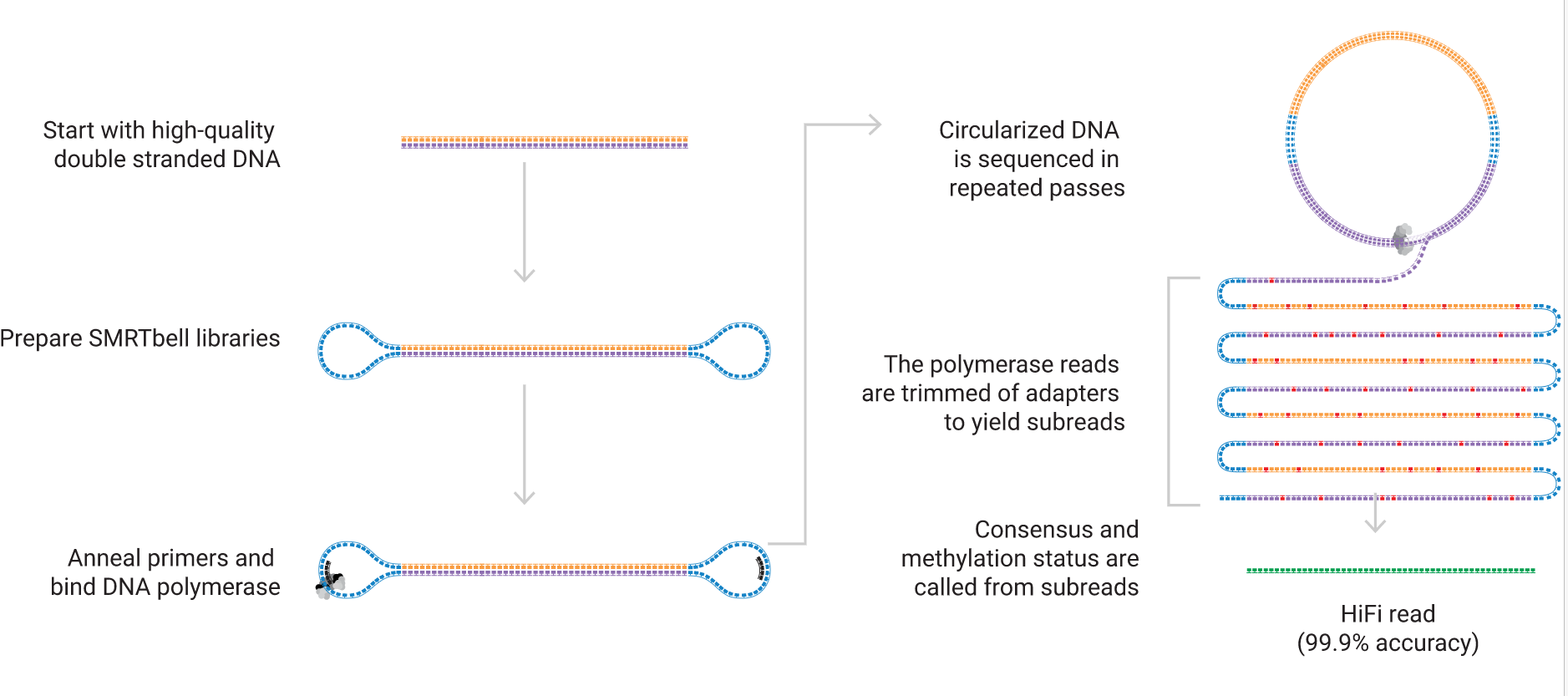
Image courtesy of PacBio (Pacific Biosciences)
Contact for Genomics and Bioinformatics Consultation
Details about sample preparation, library prep types, and application are all listed on this page. For more information or assistance contact us at ggbc@uga.edu
Helpful Links Related to PacBio Sample Preparation
PacBio Nanobind HMW DNA extraction overview
Technical-Note-Preparing-DNA-for-PacBio-HiFi-Sequencing-Extraction-and-Quality-Control
High-quality, high-molecular-weight genomic DNA is crucial for obtaining long read lengths and optimal sequencing performance. The SMRT library preparation process does not utilize amplification techniques and resulting library molecules are used directly as templates for the sequencing process. The quality of the DNA and RNA starting material will directly impact the extent of sequencing success or failure. Any unrepaired or irreversible DNA damage present in the input material (e.g., interstrand crosslinks, nicks, etc.) or contaminants will result in impaired performance in the system.
Upon receiving your DNA sample, a QC of Qubit, Nanodrop, and Fragment Analyzer is performed to ensure the DNA meets our concentration, purity, and size requirements, respectively.
Ideally, the 260/230 should be within the 2.0-2.2 range, and the 260/280 within 1.8-2.0 range. Initial DNA/RNA quality is critical for a successful run.
If you are submitting 24 or more samples, please submit samples in a 96-well plate and include an Excel file with the sample layout in your order.
PLEASE USE V BOTTOM PLATES DESIGNED FOR MOLECULAR BIOLOGY AND NOT FLAT BOTTOM PLATES DESIGNED FOR OTHER PURPOSES.
If your samples do not meet the requirements in the table below, please contact us to discuss potential options for processing your project.
PacBio Library Prep
| Application | Library Type | Description | Multiplexing | Mass Requirement | Volume Requirement | Other Requirements |
|---|---|---|---|---|---|---|
| Whole Genome Sequencing | De Novo Assembly - HiFi Reads | Produce reference quality assemblies for genomes | _ | 1 ug/ Gb of genome | ≥50 uL | 50% ≥ 30 kb & 90% ≥ 10 kb |
| Variant Detection | With 2 SMRT Cells 8M, Call SNVs, InDels, and SVs in a 3 Gb genome | _ | 1 ug/ Gb of genome | ≥50 uL | 50% ≥ 30 kb & 90% ≥ 10 kb | |
| De Novo Assembly - for Low DNA Input | Produce reference quality assemblies for genomes up to 1 Gb. Multiplex up to 2 small genomes on the Sequel II System | two 600-Mb genomes | >400 ng per 1 Gb genome size (single-sample), >300 ng per 600 mb genome size (2-plex) | ≥50 uL | 50% ≥ 30 kb & 90% ≥ 10 kb | |
| Microbial De Novo Assembly | Sequence up to 96 microbes | up to 96 microbes | >300 ng per microbe | ≥50 uL | 90% ≥ 7 kb | |
| De Novo Assembly and Variant Detection - for UltraLow DNA Input | Produce reference quality assemblies for genomes up to 500 Mb | _ | 5-20 ng per 500 Mb genome size | ≥50 uL | Majority of gDNA >20 kb | |
| Viral Sequencing | HiFiViral SARS-CoV-2 | This procedure captures the SARSCoV2 genome with tiled molecular inversion probes that create overlapping amplicons resulting in comprehensive sequence coverage | up to 384 SARS-CoV-2 samples | >10,000 copies of SARS-CoV-2 RNA per sample | ≥10 uL | _ |
| Adeno-associated virus (AAV) | Generate long and accurateHiFi reads, which can sequence an AAV genome from ITR to ITR, revealing otherwise difficult-to-detect issues | up to 24 AAV samples | ≥ 1 µg of AAV DNA per SMRT Cell, or per sample amounts should be 1 µg/ number of samples | ≥50 uL | _ | |
| RNA Sequencing | Iso-Seq Method | Characterize alternative splicing/annotate a genome with full length transcripts | up to 12 Iso-Seq samples/SMRT Cell 8M for genome annotation | 300 ng total RNA | ≥10 uL | RIN (RNA integrity number) ≥7.0 |
| MAS-Seq for 10x Single Cell | Identify cell type-specific isoforms and take advantage of long and accurate HiFi read lengths to increase single-cell Iso-Seq throughput by 16-fold through concatenated arrays of single-cell cDNA molecules | _ | 15 ng per library or 60-75 ng per library | ≥15 uL | Optimal range of 3,000-10,000 target cell recovery from the 10x Chromium 3’ single cell workflow | |
| Metagenomics | Full-length 16S rRNA Sequencing | Multiplex up to 192 samples to provide strain level resolution | up to 192 samples | 1000 ng of pooled 16S samples | ≥50 uL for a pool | _ |
| Shotgun Metagenomic Profiling or Assembly | Generate near-complette assemblies of high-complexity sample(s) (e.g. gut microbiome) | profile up to 48 communities, assemble up to 4 communities | >300 ng per sample | ≥50 uL | 90% ≥ 7 kb | |
| Targeted Sequencing | Amplicon Sequencing | HiFi sequencing can span your amplicon in a single read, giving unambiguous haplotype resolution through direct phasing | up to 1,000+ samples | Total input DNA per SMRT Cell 8M: 300 ng for <3 kb, 500 ng for 3 - 10 kb, ≥1000 ng for ≥10 kb | ≥50 uL for a pool | _ |
Contact for Financial Inquiries and Quote Requests
Please email Kim and Elizabeth at ggbc@uga.edu, for financial inquiries or to request a quote. Be as specific as possible, so that they can more quickly assist you.
A Note About Single SMRTcell Projects:
SMRTcell sequencing results depend on the final loading concentration of the library. Due to the nature of the technology, optimal concentrations can vary greatly between libraries and PacBio recommends running 2 titration SMRTcells to find the optimal concentration before running production SMRTcells. With this in mind, please be aware that projects requesting a single SMRTcell may have variable results. In most cases the first SMRTcell performs well and this is not an issue, but sometimes additional sequencing is needed. When this happens, the customer is expected to cover the cost of additional sequencing.
PacBio Table
| PacBio Service | Description | UGA Fee | Non-UGA Fee | Commercial Fee |
|---|---|---|---|---|
| HMW DNA or RNA Extraction | ||||
| DNA or RNA QC prior to extraction/purification | Volume, Concentration (ng/ul) and DNA Size | Inquire | Inquire | Inquire |
| HMW DNA extraction from Plant leaf tissue | High molecular Weight DNA extraction suitable for HiFi library preparation | Inquire | Inquire | Inquire |
| RNA preparation | RNA preparation from frozen tissue | Inquire | Inquire | Inquire |
| Library Preparation | ||||
| CCExpress Library (HiFi) Indexed | 10 µg HMW DNA minimum required | Inquire | Inquire | Inquire |
| IsoSeq Express | Express IsoSeq library sized with beads | Inquire | Inquire | Inquire |
| Sequencing | ||||
| Revio 24-h movie time | IsoSeq and short amplicons not currently supported | Inquire | Inquire | Inquire |
| Sequel II 30-h movie time | IsoSeq and short amplicons are supported | Inquire | Inquire | Inquire |
| Data Analysis | ||||
| Data Analysis | See https://dna.uga.edu/bioinformatics | Inquire | Inquire | Inquire |
The standard output files from the PacBio sequencing run are:
| Raw subreads | CCS (HiFi) reads |
| *.baz2bam_1.log | ccs_processing.report.json |
| *.scraps.bam | ccs.report.csv.zip |
| *.scraps.bam.pbi | ccs.report.json |
| *.sts.xml | ccs_tasks_report.json |
| *.subreads.bam | ccs_zmws.json.gz |
| *.subreads.bam.pbi | final.consensusreadset.xml |
| *.subreadset.xml | *.hifi_reads.bam |
| *.transferdone | *.hifi_reads.fasta.gz |
| tmp-file*.txt | *.hifi_reads.fastq.gz |
| reads.bam |
We will be sending you the following files:
| Raw subreads | CCS (HiFi) reads |
| *.baz2bam_1.log | ccs.report.csv.zip |
| *.sts.xml | final.consensusreadset.xml |
| *.subreads.bam | *.hifi_reads.bam |
| *.subreads.bam.pbi | *.hifi_reads.fasta.gz |
| *.hifi_reads.fastq.gz | |
| reads.bam |
For multiplexed runs, you will be receiving both raw and demultiplexed data.
GGBC uses Globus to transfer the data. We ask that you please confirm when you received and downloaded the data. Once your download link expires, your data is deleted on our end so please contact us beforehand if you have issues downloading the data.
- Whole Genome Sequencing:
- Targeted Sequencing:
- RNA Sequencing:
- Epigenetics Sequencing:
- Metagenome Sequencing